Biologists are hard at work on a fifteen-year project to sequence the human genome. This pattern of nucleotides encodes all the information necessary to build the proteins that we are built of. This project has already had an enormous impact on medicine and molecular biology.
Algorists have become interested in the human genome project as well, for several reasons:
My particular interest in computational biology has revolved around a recently proposed but algorithmically intensive technique for DNA sequencing called sequencing by hybridization (SBH) [CK94, PL94]. The traditional sequencing by hybridization procedure attaches a set of probes to an array, forming a sequencing chip. Each of these probes determines whether or not the probe string occurs as a substring of the DNA target. The target DNA can now be sequenced based on the constraints of which strings are and are not substrings of the target.
One problem with SBH is that enormous arrays (say ![]() strings)
are necessary to sequence relatively short pieces of DNA (typically
about 200 base pairs long).
The major reason is that all of these
strings)
are necessary to sequence relatively short pieces of DNA (typically
about 200 base pairs long).
The major reason is that all of these ![]() probes are made at the
same time.
If you want to look up a name in the telephone book but are only allowed
to consult the book once, you must copy down every single name from the
book at that time.
But if you are allowed to ask ``is the name before Mendoza?'' and wait
for the answer before asking your next question, you can use binary
search to greatly reduce your total effort.
probes are made at the
same time.
If you want to look up a name in the telephone book but are only allowed
to consult the book once, you must copy down every single name from the
book at that time.
But if you are allowed to ask ``is the name before Mendoza?'' and wait
for the answer before asking your next question, you can use binary
search to greatly reduce your total effort.
We were convinced that using several small arrays would be more efficient than using one big array. We even had theory to justify our technique, but biologists aren't very inclined to believe theory. They demand experiments for proof. Hence we had to implement our algorithms and use simulation to prove that they worked.
So much for motivation. The rest of this tale will demonstrate the impact that clever data structures can have on a string processing application.
Our technique involved identifying all the strings of length 2k that are possible substrings of an unknown string S, given that we know all length k substrings of S. For example, suppose we know that AC, CA, and CC are the only length-2 substrings of S. It is certainly possible that ACCA is a substring of S, since the center substring is one of our possibilities. However, CAAC cannot be a substring of S, since we know that AA is not a substring of S. We needed to find a fast algorithm to construct all the consistent length-2k strings, since S could be very long.

Figure: The concatentation of two fragments can be in S only
if all subfragments are
The simplest algorithm to build the 2k strings would be to concatenate
all ![]() pairs of k-strings together, and then for each pair to
make sure that all (k-1) length-k substrings spanning the boundary of the
concatenation were in fact substrings, as shown in Figure
pairs of k-strings together, and then for each pair to
make sure that all (k-1) length-k substrings spanning the boundary of the
concatenation were in fact substrings, as shown in Figure ![]() .
For example, the nine possible concatenations of AC, CA, and CC
are ACAC, ACCA, ACCC, CAAC, CACA, CACC, CCAC, CCCA,
and CCCC.
Only CAAC can be eliminated because of the absence of AA.
.
For example, the nine possible concatenations of AC, CA, and CC
are ACAC, ACCA, ACCC, CAAC, CACA, CACC, CCAC, CCCA,
and CCCC.
Only CAAC can be eliminated because of the absence of AA.
We needed a fast way of testing whether each of the k-1
substrings straddling the concatenation was a member of
our dictionary of permissible k-strings.
The time it takes to do this depends upon which kind of data structure
we use to maintain this dictionary.
With a binary search tree, we could find the correct string
within ![]() comparisons, where each comparison involved testing which of
two length-k strings appeared first in alphabetical order.
Since each such comparison could require testing k pairs of characters,
the total time using a binary search tree would be
comparisons, where each comparison involved testing which of
two length-k strings appeared first in alphabetical order.
Since each such comparison could require testing k pairs of characters,
the total time using a binary search tree would be ![]() .
.
That seemed pretty good. So my graduate student Dimitris Margaritis implemented a binary search tree data structure for our implementation. It worked great up until the moment we ran it.
``I've tried the fastest computer in our department, but our program is too slow,'' Dimitris complained. ``It takes forever on strings of length only 2,000 characters. We will never get up to 50,000.''
For interactive SBH to be competitive as a sequencing method, we had to be able to sequence long fragments of DNA, ideally over 50 kilobases in length. If we couldn't speed up the program, we would be in the embarrassing position of having a biological technique invented by computer scientists fail because the computations took too long.
We profiled our program and discovered that almost all the time was
spent searching in this data structure, which was no surprise.
For each of the ![]() possible concatenations, we did this
k-1 times.
We needed a faster dictionary data structure, since search was the innermost
operation in such a deep loop.
possible concatenations, we did this
k-1 times.
We needed a faster dictionary data structure, since search was the innermost
operation in such a deep loop.
``What about using a hash table?'' I suggested.
``If we do it right, it should take O(k) time to hash a k-character
string and look it up in our table.
That should knock off a factor of ![]() , which will mean something
when
, which will mean something
when ![]() 2,000.''
2,000.''
Dimitris went back and implemented a hash table implementation for our dictionary. Again, it worked great up until the moment we ran it.
``Our program is still too slow,'' Dimitris complained. ``Sure, it is now about ten times faster on strings of length 2,000. So now we can get up to about 4,000 characters. Big deal. We will never get up to 50,000.''
``We should have expected only a factor ten speedup,'' I mused.
``After all, ![]() .
We need a faster data structure to search in our dictionary of strings.''
.
We need a faster data structure to search in our dictionary of strings.''
``But what can be faster than a hash table?'' Dimitris countered. ``To look up a k-character string, you must read all k characters. Our hash table already does O(k) searching.''
``Sure, it takes k comparisons to test the first substring. But maybe we can do better on the second test. Remember where our dictionary queries are coming from. When we concatenate ABCD with EFGH, we are first testing whether BCDE is in the dictionary, then CDEF. These strings differ from each other by only one character. We should be able to exploit this so that each subsequent test takes constant time to perform....''
``We can't do that with a hash table,'' Dimitris observed. ``The second key is not going to be anywhere near the first in the table. A binary search tree won't help, either. Since the keys ABCD and BCDE differ according to the first character, the two strings will be in different parts of the tree.''
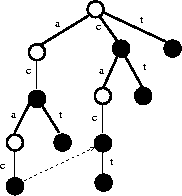
Figure: Suffix tree on ACAC and CACT, with the pointer to the suffix of ACAC
``But we can use a suffix tree to do this,'' I countered.
``A suffix tree is a trie containing all the suffixes of a given set of strings.
For example, the suffixes of ACAC are ![]() .
Coupled with suffixes of string CACT, we get the suffix tree of
Figure
.
Coupled with suffixes of string CACT, we get the suffix tree of
Figure ![]() .
By following a pointer from ACAC to its longest proper suffix CAC,
we get to the right place to test whether CACT is in our set of strings.
One character comparison is all we need to do from there.''
.
By following a pointer from ACAC to its longest proper suffix CAC,
we get to the right place to test whether CACT is in our set of strings.
One character comparison is all we need to do from there.''
Suffix trees are amazing data structures,
discussed in considerably more detail in Section ![]() .
Dimitris did some reading about them, then built a nice suffix tree
implementation for our dictionary.
Once again, it worked great up until the moment we ran it.
.
Dimitris did some reading about them, then built a nice suffix tree
implementation for our dictionary.
Once again, it worked great up until the moment we ran it.
``Now our program is faster, but it runs out of memory,'' Dimitris complained.
``And this on a 128 megabyte machine with 400 megabytes virtual memory!
The suffix tree builds a path of length k for each suffix of length
k, so all told there can be ![]() nodes in the tree.
It crashes when we go beyond 2,000 characters.
We will never get up to strings with 50,000 characters.''
nodes in the tree.
It crashes when we go beyond 2,000 characters.
We will never get up to strings with 50,000 characters.''
I wasn't yet ready to give up.
``There is a way around the space problem, by using compressed suffix trees,''
I recalled.
``Instead of explicitly representing long paths of character nodes,
we can refer back
to the original string.''
Compressed suffix trees always take linear space, as
described in Section ![]() .
.
Dimitris went back one last time and implemented the compressed suffix
tree data structure.
Now it worked great!
As shown in Figure ![]() , we ran our simulation for strings
of length
n= 65,536 on a SPARCstation 20 without incident.
Our results, reported in [MS95a], showed that interactive SBH
could be a very efficient sequencing technique.
Based on these simulations, we were able to arouse interest in our
technique from biologists.
Making the actual wet laboratory experiments feasible provided another
computational challenge, which is
reported in Section
, we ran our simulation for strings
of length
n= 65,536 on a SPARCstation 20 without incident.
Our results, reported in [MS95a], showed that interactive SBH
could be a very efficient sequencing technique.
Based on these simulations, we were able to arouse interest in our
technique from biologists.
Making the actual wet laboratory experiments feasible provided another
computational challenge, which is
reported in Section ![]() .
.
The take home lessons for programmers from Figure
![]() should be apparent.
We isolated a single operation (dictionary string search) that was being
performed repeatedly and optimized the data structure we used to
support it.
We started with a simple implementation (binary search trees) in the hopes
that it would suffice, and then used profiling to reveal the trouble when it
didn't.
When an improved dictionary structure still did not suffice,
we looked deeper into
what kinds of queries we were performing, so that
we could identify an even better
data structure.
Finally, we didn't give up until we had achieved the level of performance
we needed.
In algorithms, as in life, persistence usually pays off.
should be apparent.
We isolated a single operation (dictionary string search) that was being
performed repeatedly and optimized the data structure we used to
support it.
We started with a simple implementation (binary search trees) in the hopes
that it would suffice, and then used profiling to reveal the trouble when it
didn't.
When an improved dictionary structure still did not suffice,
we looked deeper into
what kinds of queries we were performing, so that
we could identify an even better
data structure.
Finally, we didn't give up until we had achieved the level of performance
we needed.
In algorithms, as in life, persistence usually pays off.
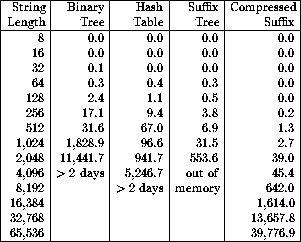
Figure: Run times (in seconds) for the SBH simulation using various data structures